


2020-02-02 by Quick Biology Inc.
In human genes, more than 95% of multi-exon genes undergo pre-mRNA alternative splicing. “One gene, more spliced isoforms” ------ this strategy is the most important contributor to protein diversity. However, researchers debate that in the transcriptome, ~ 30% genes have antisense transcripts, big parts of isoforms may be just mis-spliced, they are degraded through NMD (Nonsense-mediated Decay), cannot produce proteins. Some researchers put forward the new idea that different isoforms can be functioned in RNA level as miRNA sponge (ref1), they are not junk of transcription machinery. Scientists think questions at different angles, it indeed tells us it is a big challenge to prove novel spliced isoforms can finally produce a functional protein.
In Genome Research, Kristen Lynch Lab at the University of Pennsylvania studies which mature mRNA isoforms are successfully translated into protein (ref2). They used high throughput RNA-sequencing, found spliced junction reads. By combining mass spectrometry-based proteomics data and RefSeq databases, they improve the identification of splicing-derived proteoforms by 28% compared with the use of the SWISS-PROT database alone (Fig.1). By performing this new method, authors tracked changes in the transcriptome and proteome induced by T-cell stimulation (Fig. 2).
Figure 1: Identification of local splice variations (LSVs) and alternative junctions (AJs). (A) Workflow for identifying significantly used AJs in mRNA by identifying LSVs containing two or more exon junctions. (B) Distribution of AJ usage in unstimulated and stimulated conditions. (C) Distribution of LSVs with two or more AJs throughout transcript regions, namely, the coding sequence (CDS) and untranslated regions (UTRs). (D) Workflow for generating MAJIQ custom protein database using AJs from A.
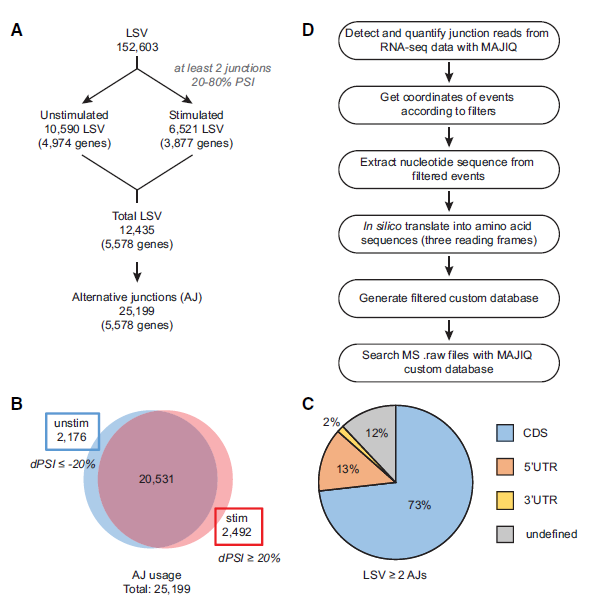
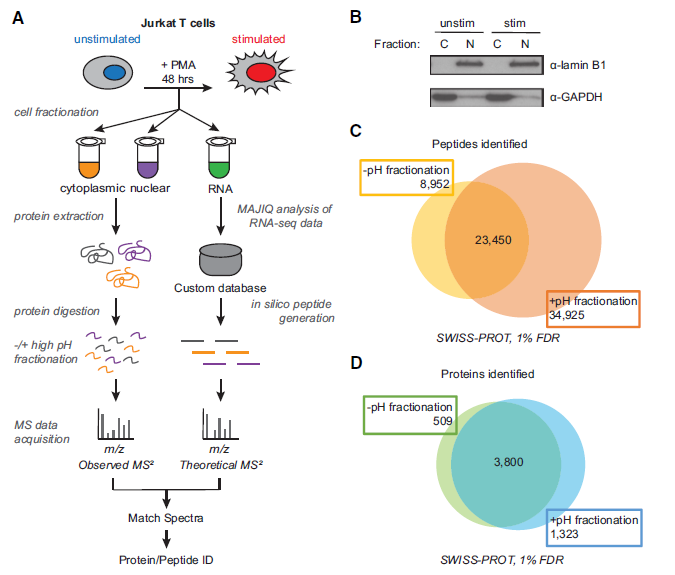
Figure 2: Increased protein and peptide identification achieved by high pH peptide fractionation. (A) Sample processing workflow used for integration of mass spectrometry (MS)–based proteomics and RNA-seq data. (B) Validation of subcellular fractionation by western blot using antibodies for GAPDH and lamin B1. (C) Number of peptides identified −/+ high pH peptide fractionation at 1% false-discovery rate (FDR). (D) Number of proteins identified −/+ high pH peptide fractionation.
Quick Biology is an expert in the transcriptome, genomes, and proteomics. Find More at Quick Biology.
Ref:
1. Lu, Z. X. Gene self-control: when pre-mRNA splicing variants become competing endogenous RNAs. Front. Genet. (2014). doi:10.3389/fgene.2014.00405
2. Agosto, L. M. et al. Deep profiling and custom databases improve detection of proteoforms generated by alternative splicing. Genome Res. 1–10 (2019). doi:10.1101/gr.248435.119.