


2020-12-28 By Quick Biology
Chronic lymphocytic leukemia (CLL) is a disease that remains incurable by conventional chemotherapy. SF3B1 is a splicing factor, a core component of the U2 snRNP of the spliceosome, and associates with the U2 snRNA and branch point adenosine of the pre-mRNA. Recurrent somatic mutations in SF3B1 have been linked to CLL. In CLL, mutations in the heat-repeat domain of SF3B1 is shown to associate with poor clinical outcome. Previous whole transcript profiling mainly focused on exon level using Illumina short reads NGS platform. The extent to which full-length isoforms contribute to such disease due to splicing regulator SF3B1 mutations are not well characterized.
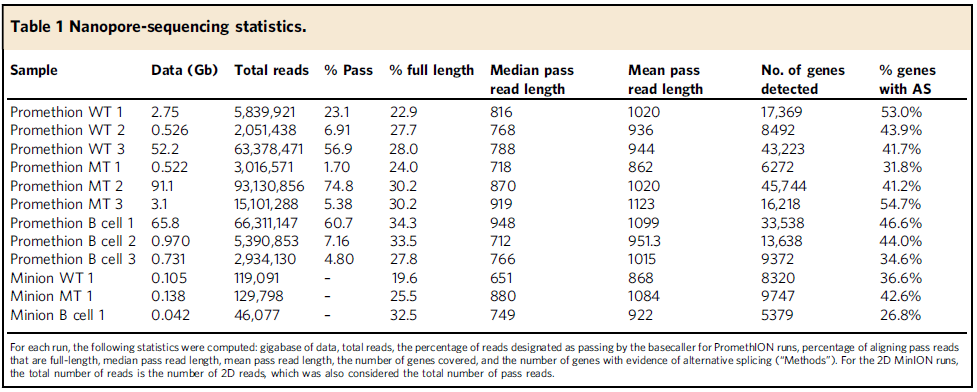
In Nature communications, Brooks Lab at UCSC (https://brookslab.soe.ucsc.edu/people/) with her collaborator Prof. Catherine Wu in Dana-Farber Cancer Institute used Nanopore sequencing for full-length cDNA from CLLL samples with and without SF3B1 mutation, as well as normal B cell samples (ref1, Fig.1). They also present FLAIR (Full-Length Alternative Isoform analysis of RNA), a computational workflow to identify high-confidence transcripts, perform differential splicing event analysis, and differential isoform analysis. Using their total of 149 million pass reads of nanopore sequencing (Table 1), they found differential 3’ splice site changes associated with SF3B1 mutation, which agreed with SF3B1 role as its core component of U2 snRNP complex binding to 3’splice site, branch point region. Interestingly, they also observed a strong downregulation of intron retention events. This full-length analysis (both Nanopore platform and FLAIR method) work allows us for better estimates of splicing dynamics in cancer research.
Figure 1: Long-read nanopore sequencing and FLAIR analysis pipeline (ref1). a RNA from primary samples across three conditions (chronic lymphocytic leukemia with and without SF3B1 mutation and normal B cells) were obtained. The RNA was prepared into 1D cDNA libraries and each sample was sequenced on a PromethION flow cell. The basecalled data were processed using the FLAIR pipeline. b The FLAIR pipeline constructs an isoform set from nanopore reads. First, reads are aligned to the genome with a spliced aligner. The sequence errors are marked in red. Next, they are splice-corrected using splice sites from either annotated introns, introns from short-read data, or both. The corrected reads are grouped by their splice junction chains and are summarized into representative isoforms (first-pass set). All reads are then reassigned to a first-pass isoform. The isoforms that surpass a supporting read threshold of 3 comprise the final high-confidence isoform set.
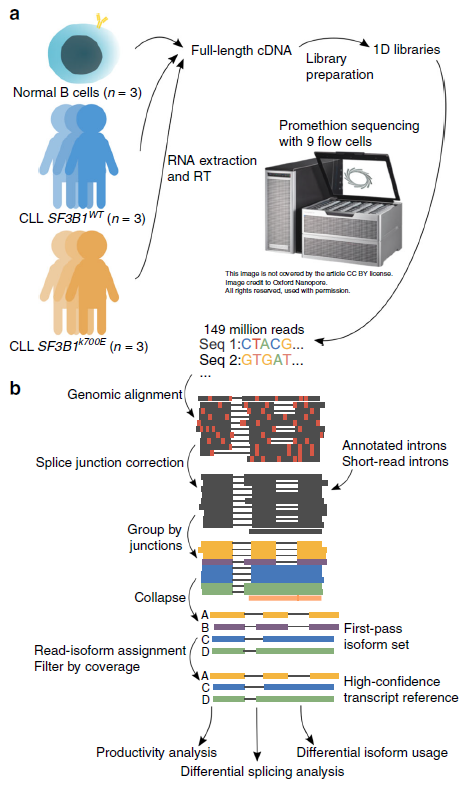
Table 1: Full-length cDNA Nanopore sequencing statistics. (Ref1)
Quick Biology can assistant you with Nanopore long-read projects such as full-length cDNA sequencing. Find More at Quick Biology.
See resource:
1. Tang, A. D. et al. Full-length transcript characterization of SF3B1 mutation in chronic lymphocytic leukemia reveals downregulation of retained introns. Nat. Commun. 11, 1–12 (2020).