


Introduction
ATAC-seq (Assay For Transposase-Accessible Chromatin Sequencing) is a fast and sensitive high-throughput sequencing method for epigenomic profiling of open chromatin, DNA-binding proteins, and nucleosome position. ATAC-seq uses hyperactive Tn5 transposase to simultaneously cut and ligate adapters for high-throughput sequencing at regions of increased accessibility. This technique will allow multidimensional assays of the regulatory landscape of chromatin with a relatively simple and fast protocol. Unlike methods such as MNase-seq, ChIP-seq, and DNase-seq which often require tens to hundreds of millions of cells as input material, ATAT-seq can be carried out with a standard sample size of 50,000 cells. Therefore, ATAC-seq is a fast and sensitive alternative to DNase-Seq for assaying genome-wide chromatin accessibility, or to MNase for assaying nucleosome positions in accessible regions of the genome.
Request an ATAC-Seq Quote, Click Here!
Sample Types:
-- human or aminal cells
-- FACS sorted cells
-- Primary cells (including T and B cells)
-- Other rare cells population
-- Tissues (frozen or sections)
Frequently Asked Questions:
1. What are the consequences of using inappropriate numbers of cells for ATAC-seq?
ATAC-seq is very robust to relatively minor variations in cell number. In general, using too many cells causes under-digestion and creates high molecular weight fragments, which is not optimal for sequencing; Using too few cells causes over-digestion of chromatin and may result in reads that can map to inaccessible regions of the genome (a source of noise).
2. What are the best ways to collect cells for ATAC-seq?
There are many methods for cell collection which may need to be optimized for ATAC-seq. In general, we found that using fixatives can reduce transposition frequency and is not recommended. Mechanical shearing can also significantly reduce signal-to-noise. To have the best data, cells should be intact and in a homogenous single cell suspension. We recommend resuspending the cells in Cryostor media (or a compatible freezing media with DMSO) and freeze the cell vials at -80°C overnight slowly in an isopropyl alcohol chamber (aka Mr. Frosty) or similar cooling device at a rate of -1°C per minute.
3. Why do we obtain many mitochondrial reads in our ATAC-seq data?
It is normal to have a fraction of reads derived from mitochondrial DNA because of their relative abundance in the cell population. It is common to observe the fraction of the mitochondrial reads vary between 10%-50% depending on the cell type, but we have optimized the protocol to control the fraction of mitochondrial reads as low as 5-15%.
4. How much sequencing depth is required for ATAC-seq?
For nucleosome mapping, paired-end sequencing is preferred. Paired-end 50-150bp cycle reads generally provide accurate alignments. For inferring differences in open chromatin within human samples, it generally requires >50,000,000 mapped reads, and for transcription factor foot-printing, it is better to have >200,000,000 mapped reads.
Workflow
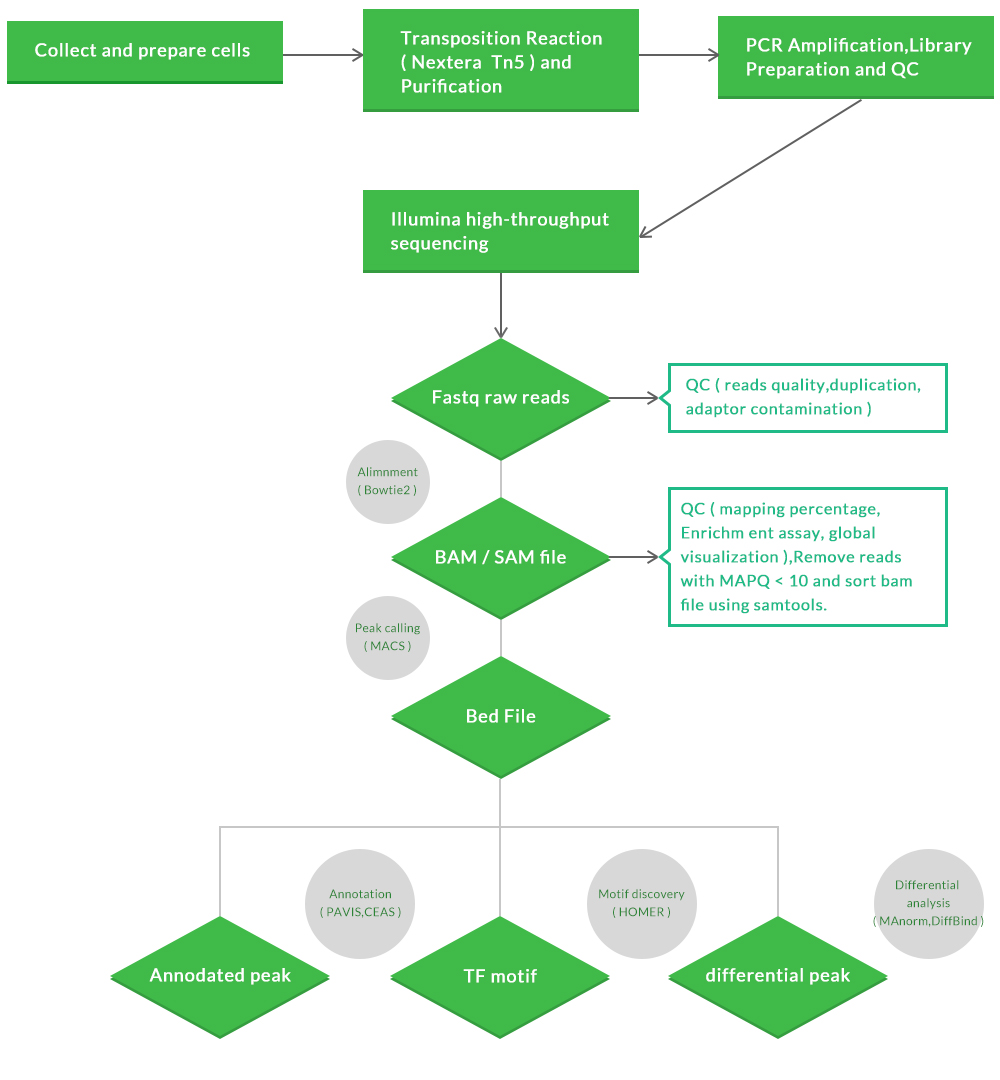
ATAC-seq sample preparation, sequencing, and data analysis workflow:
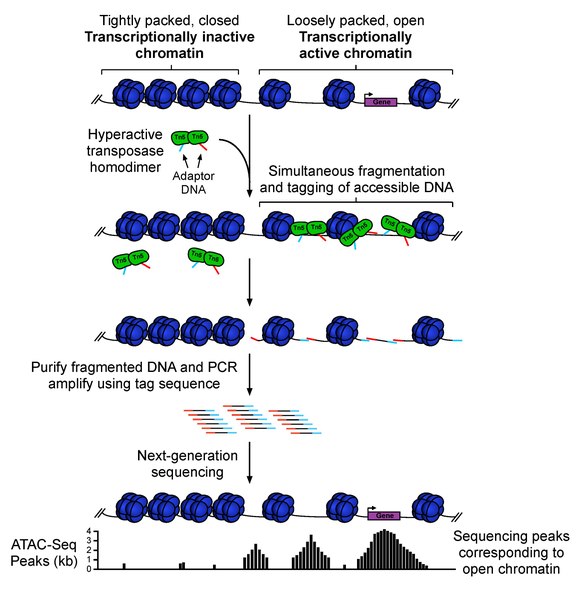
1. Collect and prepare cells for lysis
2. Transposition reaction and purification
3. PCR amplification, library preparation, and QC
4. Illumina High-throughput sequencing (> 60M total reads of Paired-end 75-150 bp recommended)
5. Bioinformatics data analysis to get useful insights
Our ATAC-seq workflow:
Sample Requirment
-- Cell number: 50,000 - 75,000 cells needed, but we recommended 200,000 to 500,000 cells (species dependent) with high viability (90-95%) to ensure high data quality.
-- Cell Collection: Cells should be intact and in a homogenous single cell suspension, fixatives can reduce transposition frequency and are not recommended.
-- Tissues: 20 - 50 mg
Please refer to Quick Biology Sample Preparation & Shipping instructions for more details.
Data Analysis
Standard Analysis:
a.Raw data QC and clean up
b.Alignment to a reference with mapping statistics
c.Peaking calling with or without control samples
d.Gene assignment and peak annotation
e.Pathway Analysis
f. Final project report (HTML) with analysis methods, publication-ready graphics, and reference
Advanced Analysis:
h.Motif analysis
i.clustering and visualization
Please download the ATAC-seq data analysis sample report here!
Turnaround Time
~3-4 weeks depending on project requirements, one additional week for data analysis